MicroRNA sequencing (miRNA-seq), a type of RNA-Seq, is the use of next-generation sequencing or massively parallel high-throughput DNA sequencing to sequence microRNAs, also called miRNAs. miRNA-seq differs from other forms of RNA-seq in that input material is often enriched for small RNAs. miRNA-seq allows researchers to examine tissue-specific expression patterns, disease associations, and isoforms of miRNAs, and to discover previously uncharacterized miRNAs. Evidence that dysregulated miRNAs play a role in diseases such as cancer[1] has positioned miRNA-seq to potentially become an important tool in the future for diagnostics and prognostics as costs continue to decrease.[2] Like other miRNA profiling technologies, miRNA-Seq has both advantages (sequence-independence, coverage) and disadvantages (high cost, infrastructure requirements, run length, and potential artifacts).[3]
Introduction
MicroRNAs (miRNAs) are a family of small ribonucleic acids, 21-25 nucleotides in length, that modulate protein expression through transcript degradation, inhibition of translation, or sequestering transcripts.[4][5][6] The first miRNA to be discovered, lin-4, was found in a genetic mutagenesis screen to identify molecular elements controlling post-embryonic development of the nematode Caenorhabditis elegans.[7] The lin-4 gene encoded a 22 nucleotide RNA with conserved complementary binding sites in the 3’-untranslated region of the lin-14mRNA transcript[8] and downregulated LIN-14 protein expression.[9] miRNAs are now thought to be involved in the regulation of many developmental and biological processes, including haematopoiesis (miR-181 in Mus musculus[10]), lipid metabolism (miR-14 in Drosophila melanogaster[11]) and neuronal development (lsy-6 in Caenorhabditis elegans[12]).[6] These discoveries necessitated development of techniques able to identify and characterize miRNAs, such as miRNA-seq.
History
MicroRNA sequencing (miRNA-seq) was developed to take advantage of next-generation sequencing or massively parallel high-throughput sequencing technologies in order to find novel miRNAs and their expression profiles in a given sample. miRNA sequencing in and of itself is not a new idea, initial methods of sequencing utilized Sanger sequencing methods. Sequencing preparation involved creating libraries by cloning of DNA reverse transcribed from endogenous small RNAs of 21–25 bp size selected by column and gel electrophoresis.[13] However, this method is exhaustive in terms of time and resources, as each clone has to be individually amplified and prepared for sequencing. This method also inadvertently favors miRNAs that are highly expressed.[6] Next-generation sequencing eliminates the need for sequence specific hybridization probes required in DNA microarray analysis as well as laborious cloning methods required in the Sanger sequencing method. Additionally, next-generation sequencing platforms in the miRNA-SEQ method facilitate the sequencing of large pools of small RNAs in a single sequencing run.[14]
miRNA-seq can be performed using a variety of sequencing platforms. The first analysis of small RNAs using miRNA-seq methods examined approximately 1.4 million small RNAs from the model plant Arabidopsis thaliana using Lynx Therapeutics' Massively Parallel Signature Sequencing (MPSS) sequencing platform. This study demonstrated the potential of novel, high-throughput sequencing technologies for the study of small RNAs, and it showed that genomes generate large numbers of small RNAs with plants as particularly rich sources of small RNAs.[15] Later studies used other sequencing technologies, such as a study in C. elegans which identified 18 novel miRNA genes as well as a new class of nematode small RNAs termed 21U-RNAs.[16] Another study comparing small RNA profiles of human cervical tumours and normal tissue, utilized the Illumina (company) Genome Analyzer to identify 64 novel human miRNA genes as well as 67 differentially expressed miRNAs.[17]Applied Biosystems SOLiD sequencing platform has also been used to examine the prognostic value of miRNAs in detecting human breast cancer.[18]
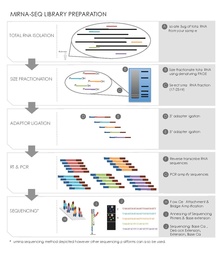
Sequence library construction can be performed using a variety of different kits depending on the high-throughput sequencing platform being employed. However, there are several common steps for small RNA sequencing preparation.[19][20]
Total RNA Isolation
In a given sample all the RNA is extracted and isolated using an isothiocyanate/phenol/chloroform (GITC/phenol) method or a commercial product such as Trizol (Invitrogen) reagent. A starting quantity of 50-100 μg total RNA, 1 g of tissue typically yields 1 mg of total RNA, is usually required for gel purification and size selection.[20] Quality control of the RNA is also measured, for example running an RNA chip on Caliper LabChipGX (Caliper Life Sciences).
Size Fractionation of small RNAs by Gel Electrophoresis
Isolated RNA is run on a denaturing polyacrylamide gel. An imaging method such as radioactive 5’-32P-labeled oligonucleotides along with a size ladder is used to identify a section of the gel containing RNA of the appropriate size, reducing the amount of material ultimately sequenced. This step does not have to be necessarily carried out before the ligation and reverse transcription steps outlined below.[19][20]
Ligation
The ligation step adds DNA adaptors to both ends of the small RNAs, which act as primer binding sites during reverse transcription and PCR amplification. An adenylated single strand DNA 3’adaptor followed by a 5’adaptor is ligated to the small RNAs using a ligating enzyme such as T4 RNA ligase2. The adaptors are also designed to capture small RNAs with a 5’ phosphate group, characteristic microRNAs, rather than RNA degradation products with a 5’ hydroxyl group.[19][20]
Reverse Transcription and PCR Amplification
This step converts the small adaptor ligated RNAs into cDNA clones used in the sequencing reaction. There are many commercial kits available that will carry out this step using some form of reverse transcriptase. PCR is then carried out to amplify the pool of cDNA sequences. Primers designed with unique nucleotide tags can also be used in this step to create ID tags in pooled library multiplex sequencing.[19][20]
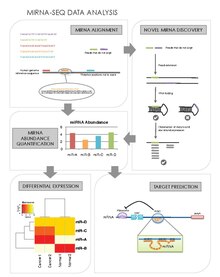
Central to miRNA-seq data analysis is the ability to 1) obtain miRNA abundance levels from sequence reads, 2) discover novel miRNAs and then be able to 3) determine the differentially expressed miRNA and their 4) associated mRNA gene targets.
miRNA-seq Data Analysis
miRNA Alignment & Abundance Quantification
miRNAs may be preferentially expressed in certain cell types, tissues, stages of development, or in particular disease states such as cancer.[1] Since deep sequencing (miRNA-seq) generates millions of reads from a given sample, it allows us to profile miRNAs; whether it may be by quantifying their absolute abundance, to discover their variants (known as isomirs[25]) Note that given that the average length of sequence reads are longer than the average miRNA (17-25 nt), the 3’ and 5’ ends of the miRNA should be found on the same read.
There are several miRNA abundance quantification algorithms.[21][26] Their general steps are as follows:[27]
After sequencing, the raw sequence reads are filtered based on quality. The adaptor sequences are also trimmed off the raw sequence reads.
The resulting reads are then formatted into a fasta file where the copy number and sequence is recorded for each unique tag.
Sequences that may represent E. Coli contamination are identified by a BLAST search against an E. Coli database and are removed from analysis.
Each of the remaining sequences are aligned against a miRNA sequence database (such as miRBase[28]) In order to account for imperfect DICER processing, a 6nt overhang on the 3’ end, and 3nt on the 5’ end are allowed.
The reads that do not align to the miRNA database are then loosely aligned to miRNA precursors to detect miRNAs that might carry mutations or those that have gone through RNA editing.
The read counts for each miRNA are then normalized to the total number of mapped miRNAs to report the abundance of each miRNA.
Novel miRNA Discovery
Another advantage of miRNA-seq is that it allows the discovery of novel miRNAs that may have eluded traditional screening and profiling methods.[27] There are several novel miRNA discovery algorithms. Their general steps are as follows:
Obtain reads that did not align to known miRNA sequences, and map them to the genome.
RNA Folding Method
For the miRNA sequences were an exact match is found, obtain the genomic sequence including ~100bp of flanking sequence on either side, and run the RNA through RNA folding software such as the Vienna package.[29]
Folded sequences that lie on one arm of the miRNA hairpin and have a minimum free energy of less than ~25kcal/mol are shortlisted as putative miRNA.
The shortlisted sequences are trimmed down to include only the possible precursor sequence and are then refolded to ensure that the precursor was not artificially stabilized by neighbouring sequences.
The resulting folded sequences are considered novel miRNAs if the miRNA sequence falls within one arm of the hairpin, and are highly conserved between species.
Novel miRNA sequences are identified based on the characteristic expression pattern that they display due to DICER processing: higher expression of the mature miRNA over the star strand and loop sequences.
Differential Expression Analysis
After the abundances of miRNAs are quantified for each sample, their expression levels can be compared between samples. One would then be able to identify miRNA that are preferentially expressed that particular time points, or in particular tissues or disease states. After normalizing for the number of mapped reads between samples, one can use a host of statistical tests (like those used in gene expression profiling) to determine differential expression
Target Prediction
Identifying a miRNA's mRNA targets will provide an understanding of the genes or networks of genes whose expression they regulate.[31] Public databases provide predictions of miRNA targets. But to better distinguish true positive predictions from false positive predictions, miRNA-seq data can be integrated to mRNA-seq data to observe for miRNA:mRNA functional pairs. RNA22,[32]TargetScan,[33][34][35][36][37][38] miRanda,[39] and PicTar[40] are software designed for this purpose. A list of prediction software is given here.
The general steps are:
Determine miRNA:mRNA binding pairs, complementarity between the miRNA sequences at the 3’-UTR of the mRNA sequence is identified.
Determine the degree of conservation of miRNA:mRNA binding pairs across species. Typically, more highly binding pairs are less likely to be false positives of prediction.
Observe for evidence of miRNA targeting in mRNA-seq or protein expression data: where the miRNA expression is high, the gene and protein expression of its target gene should be low.
Target Validation for Cleaved mRNA Targets
Many miRNAs function to direct cleavage of their mRNA targets; this is particularly true in plants, and thus high-throughput sequencing methods have been developed to take advantage of this property of miRNAs by sequencing the uncapped 3' ends of cleaved or degraded mRNAs. These methods are known as Degradome sequencing or PARE.[41][42] Validation of target cleavage in specific mRNAs is typically performed using a modified version of 5' Rapid Amplification of cDNA Ends with a gene-specific primer.
Applications
Identification of Novel miRNAs
miRNA-seq has revealed novel miRNAs that were previously eluded in traditional miRNA profiling methods. Examples of such findings are in embryonic stem cells,[25] chicken embryos,[43] acute lymphoblastic leukaemia,[44] diffuse large b-cell lymphoma and b-cells,[45] acute myeloid leukemia,[46] and lung cancer.[47]
Disease biomarkers
Micro RNAs are important regulators of almost all cellular processes such as survival, proliferation, and differentiation. Consequently, it is not unexpected that miRNAs are involved in various aspects of cancer through the regulation of onco- and tumor suppressor gene expression. In combination with the development of high-throughput profiling methods, miRNAs have been identified as biomarkers for cancer classification, response to therapy, and prognosis.[48] Additionally, because miRNAs regulate gene expression they can also reveal perturbations in important regulatory networks that may be driving a particular disorder.[48] Several applications of miRNAs as biomarkers and predictors of disease are given below.
Table 1: Cancer subtypes distinguished by microRNAs
αThis is not a comprehensive list of miRNAs involved with these malignancies.
Comparison With Other Methods of miRNA Profiling
The disadvantages of using miRNA-seq over other methods of miRNA profiling are that it is more expensive, generally requires a larger amount of total RNA, involves extensive amplification, and is more time-consuming than microarray and qPCR methods.[3] As well, miRNA-seq library preparation methods seem to have systematic preferential representation of the miRNA complement, and this prevents accurate determination of miRNA abundance.[64] At the same time, the approach is hybridization independent and therefore does not require a priori sequence information. Because of this, one can obtain sequences of novel miRNAs and miRNA isoforms (isoMirs), distinguish sequentially similar miRNAs, and identify point mutations.[65]
^Sandhu, S.; Garzon, R. (2011). "Potential Applications of MicroRNAs in Cancer Diagnosis, Prognosis, and Treatment". Semin Oncol. 38 (6): 781–787. doi:10.1053/j.seminoncol.2011.08.007. PMID22082764.
^Hofacker, I. L.; Fontana, W.; Stadler, P. F.; Bonhoeffer, L. S.; Tacker, M.; Schuster, P. (1994). "Fast folding and comparison of RNA secondary structures". Monatshefte für Chemie. 125 (2): 167–188. doi:10.1007/BF00818163. ISSN0026-9247. S2CID19344304.
^Krek, A. Identification of microRNA targets. DAI-B 70/07, (2010).
^German MA, Pillay M, Jeong DH, Hetawal A, Luo S, Janardhanan P, Kannan V, Rymarquis LA, Nobuta K, German R, De Paoli E, Lu C, Schroth G, Meyers BC, Green PJ (2008). "Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends". Nat. Biotechnol. 26 (8): 941–946. doi:10.1038/nbt1417. PMID18542052. S2CID13187064.
^Marcucci, Guido; Radmacher, Michael D.; Maharry, Kati; Mrózek, Krzysztof; Ruppert, Amy S.; Paschka, Peter; Vukosavljevic, Tamara; Whitman, Susan P.; Baldus, Claudia D.; Langer, Christian; Liu, Chang-Gong; Carroll, Andrew J.; Powell, Bayard L.; Garzon, Ramiro; Croce, Carlo M.; Kolitz, Jonathan E.; Caligiuri, Michael A.; Larson, Richard A.; Bloomfield, Clara D. (2008). "MicroRNA Expression in Cytogenetically Normal Acute Myeloid Leukemia". New England Journal of Medicine. 358 (18): 1919–1928. doi:10.1056/NEJMoa074256. ISSN0028-4793. PMID18450603.
^Calin, George Adrian; Ferracin, Manuela; Cimmino, Amelia; Di Leva, Gianpiero; Shimizu, Masayoshi; Wojcik, Sylwia E.; Iorio, Marilena V.; Visone, Rosa; Sever, Nurettin Ilfer; Fabbri, Muller; Iuliano, Rodolfo; Palumbo, Tiziana; Pichiorri, Flavia; Roldo, Claudia; Garzon, Ramiro; Sevignani, Cinzia; Rassenti, Laura; Alder, Hansjuerg; Volinia, Stefano; Liu, Chang-gong; Kipps, Thomas J.; Negrini, Massimo; Croce, Carlo M. (2005). "A MicroRNA Signature Associated with Prognosis and Progression in Chronic Lymphocytic Leukemia". New England Journal of Medicine. 353 (17): 1793–1801. doi:10.1056/NEJMoa050995. ISSN0028-4793. PMID16251535.